Analysis¶
This sections explains how OncodriveFML compute the scores for the observed mutations and how mutations are simulated.
The analysis is done for each element independently. The same number of observed mutations is simulated within the element, taking only the positions indicated in the regions file.
Observed¶
Single Nucleotide Polymorphism (SNP)¶
SNP mutations are the simplest to compute. To score them, OncodriveFML get the score for the corresponding alteration in the position of the mutation.
If there is not a score for that particular change, the mutation is ignored [1].
Multi Nucleotide Polymorphism (MNP)¶
MNP mutations are considered as set of SNPs. The observed value is the maximum value of all the changes produced by the MNP.
MNPs are ignored [1] when none of the changes it introduces has a score.
Insertion or deletion (INDEL)¶
Indels are scored in two different ways: as substitutions or as stops.
- As substitutions
Indels that fall in non-coding regions or in-frame indels in coding regions are considered as a set of substitutions. Similarly to MNP mutations, the changes produced by the indel are computed as a set of SNPs mutation and OncodriveFML assigns the indel the maximum score of those changes. In an insertion, the reference genome is compared with the indel. In a deletion, the reference genome is compared with itself but shifted a number of position equal to the length of the indel. Only the changes produced in the length of the indel are considered.
Note
If none of the changes produced by the indel has a score, the indel is ignored [1].
- As stops
Indels can be scored as stops in the analysis of coding regions and if their length is not a multiple of 3. In coding regions, a frameshift indel might cause, somewhere in the gene, a stop. This is why OncodriveFML can use this approach. The way OncodriveFML scores this type of indels is taking all the stop scores [2] in the gene under analysis and applying a user defined function to them. In some cases, OncodriveFML can infer a value for the scores of the stops using the mean score of all mutations in the gene. See the configuration of indel section for further information.
Note
This option needs to be manually set up in the using the configuration file.
Indels with a length higher than 20 nucleotides are ignored [1].
Simulated¶
The same number of mutations that are observed and have a score are simulated.
To perform the simulation two arrays are computed:
- One contains the scores of all possible changes to be simulated.
- The other array contains the probabilities of each of those changes.
Using the probability array, a random sampling of the scores array is done to obtain the simulated scores.
Probabilities¶
The probability array is computed taking into account different parameters.
If only substituions are simulated, either because the analysis excludes indels or because they are simulated as substitutions, the probabilities are:
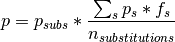
where s represents each of the signatures found in the gene in the observed mutations,
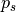 is the probability of a particular mutation to occur given the
is the probability of a particular mutation to occur given the s signature,
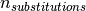 is the total number of substitutions,
and
is the total number of substitutions,
and 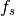 is the relative frequency of a particular signature
is the relative frequency of a particular signature s in the gene.
However, if you are not using any signature (see singature configuration):
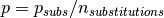
where 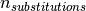 is the amount of substitutions in the gene.
is the amount of substitutions in the gene.
However, if you configure indels to be analysed as stops things are slightly more complex. Substitution are simulated as explained above, as well as in frame indels. However, there is also a chance that a the score of one stop is selected.
The probability associated to any of the stop scores is:
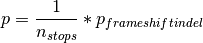
where 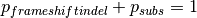 , and
, and 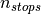 is the number of
stop scores for that gene.
is the number of
stop scores for that gene.
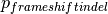 represents the probability of simulating a frameshift indel in that gene,
and
represents the probability of simulating a frameshift indel in that gene,
and  represents the probability of simulating a substitution.
represents the probability of simulating a substitution.
The probability of simulating a frameshift indel, also, depends on whether you are analysing using the whole cohort percentages or only the mutations observed in each gene.
When using exomic frameshift probabilities OncodriveFML computes how many indels you observe, and how many of those fall into the region you are analysing (which should be coding). Among the mapped indels OncodriveFML distinguishes between frameshift and in-frame indels. The ratio of frameshift indels against the total amount of mutations is used to compute
.When using the probabilities taken from the gene:
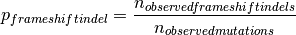
where
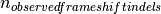 is the number of observed frameshift indels
and
is the number of observed frameshift indels
and 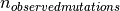 is the number of observed mutations.
is the number of observed mutations.
| [1] | (1, 2, 3, 4) When an observed mutation is ignored it means that it cannot be assigned a score, and thus it does not contribute to the observed scores and in the simulation the number of mutations simulated is one less for that region. |
| [2] | The package BgData includes the precomputed position and alteration of the stops for the HG19 genome build. OncodriveFML makes use of it. |