Signature¶
The signature is an array that assigns a probability to a single nucleotide mutation taking into account its context [1]. It represents the chance of a certain mutation to occur within a context.
Check the different options for the signature in the configuration file. In short, you can choose between not using any signature, using your own signature or computing the signature from the mutations file. Additionally, signatures can be grouped into different categories (such as the sample).
The signature is computed count all the Single Nucleotide Polymorphisms
in the input file, taking into account their context.
The counts are used to compute a frequency
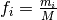 where
where 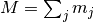 , and
, and
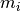 represent the number of times that the mutation
represent the number of times that the mutation
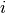 with its context [1] has been observed.
with its context [1] has been observed.
Optionally, the signature can be corrected taking into
account the frequency of trinucleotides in the
reference genome.
OncodriveFML introduces this feature because the
distribution of triplets is not expected to be constant.
When using the command line interface, OncodriveFML
does this correction automatically according to
the value passed in the flag --sequencing
(you can list all the options using the help).
Important
Signature correction is done using precomputed counts of whole genome and whole exome of HG19 reference genome.
This counts might be similar for other human genomes but ensure that correction is not done genomes of other species. Check the command line and configuration file.
More complex signatures (e.g. using only mutations that map to the regions under analysis, or normalizing by the frequency of trinucleotides in specific regions of the genome) can be computed using the bgsignature package and passed to OncodriveFML via the configuration file.
Reasoning behind the correction¶
Let’s first take the conditional probability of a mutation (with contectx [1])
to occur given the number of those triplets in the region:
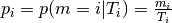 .
.
Then, the normalized frequency of the mutation is:
 .
.
The results can be adapted in case our inputs are not absolute values but relative frequencies.
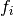 is the frequency of mutations and
is the frequency of mutations and 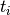 the frequency of nucleotides:
the frequency of nucleotides:
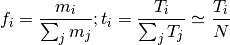
(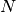 is the number of nucleotides,
is the number of nucleotides, 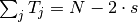 , where
, where  is the number of segments)
is the number of segments)
Then:
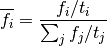
Proof:
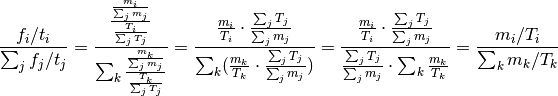
| [1] | (1, 2, 3) The context is formed by the previous and posterior nucleotides. |