Output¶
OncodriveFML generates 3 output files:
- A
.tsvwith the analysis results - A
.pngimage with the most significant genes labeled. - A
.htmlinteractive plot which can be used to search for specific genes.
Naming¶
All the 3 files generated by OncodriveFML have the same name.
They only differ in the extension.
The name given to the files is the same as the name of the
mutations file followed by -oncodrivefml and the extension.
The .tsv file¶
This tabulated file is the most important (as the others are just plots using the data in this one) and contains the results of the analysis.
In the file, the following columns can be found:
- index
- Gene ID from Ensembl
- MUTS
- number of mutations found in the dataset for that gene
- MUTS_RECURRENCE
- number of mutations that do not occur in the same position
- SAMPLES
- number of mutated samples in the gene
- P_VALUE
- times that the observed value is higher than or equal to the expected value, divided by the number of randomizations
- Q_VALUE
pvaluecorrected using the Benjamini/Hochberg correction (for samples with at least 2samples_mut)- P_VALUE_NEG
- times that the observed value is lower than or equal to the expected value, divided by the number of randomizations
- Q_VALUE_NEG
pvalue_negcorrected using the Benjamini/Hochberg correction (for samples with at least 2samples_mut)- SNP
- number of mutations that are Single Nucleotide Polymorphisms
- MNP
- number of mutations that are Multi Nucleotide Polymorphisms (two or more)
- INDELS
- number of mutations that are insertions or deletions
- SYMBOL
- HGNC Symbol
The plots¶
Both plots (.png and .html) represent the same.
They are similar to Q-Q plots
where in the Y axis the 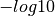 of the computed P-values are represented (sorted)
and in the X axis the of the expected P-values are reported (sorted).
of the computed P-values are represented (sorted)
and in the X axis the of the expected P-values are reported (sorted).
The expected P-values represent the null distribution: 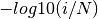 where
where ![i \in [1, N]](_images/math/0715c5c11e61175226d3add8fd735a8f17f9ed71.png) and
and N represents the number of computed
P-values.
Note
The P-values of OncodriveFML are always > 0, even when all the simulated functional impact scores are lower than the observed functional impact score. In this case, a pseudocount is added.
The genomic elements that have a lighter color in the plot are the ones for which the number of mutated sample does not reach the minimum required to perform the multiple test correction.
All the genomic regions above the red line in the plot represent those with a Q-value below 0.1. The ones between the green line and the red line are the ones with a Q-value between 0.25 and 0.1.